Input data and parameters
Input
| Analysis date: | Fri Jul 18 00:44:31 GMT 2025 |
| BAM file: | DE-2.markdup.sorted.bam |
| Counting algorithm: | uniquely-mapped-reads |
| GTF file: | genes.filtered.gtf |
| Number of bases for 5'-3' bias computation: | 100 |
| Number of transcripts for 5'-3' bias computation: | 1,000 |
| Paired-end sequencing: | yes |
| Protocol: | strand-specific-reverse |
| Sorting performed: | yes |
Summary
Reads alignment
| Number of mapped reads (left/right): | 83,069,646 / 83,046,675 |
| Number of aligned pairs (without duplicates): | 83,017,779 |
| Total number of alignments: | 181,107,831 |
| Number of secondary alignments: | 14,991,510 |
| Number of non-unique alignments: | 23,121,669 |
| Aligned to genes: | 82,091,802 |
| Ambiguous alignments: | 599,490 |
| No feature assigned: | 75,286,151 |
| Missing chromosome in annotation: | 8,719 |
| Not aligned: | 0 |
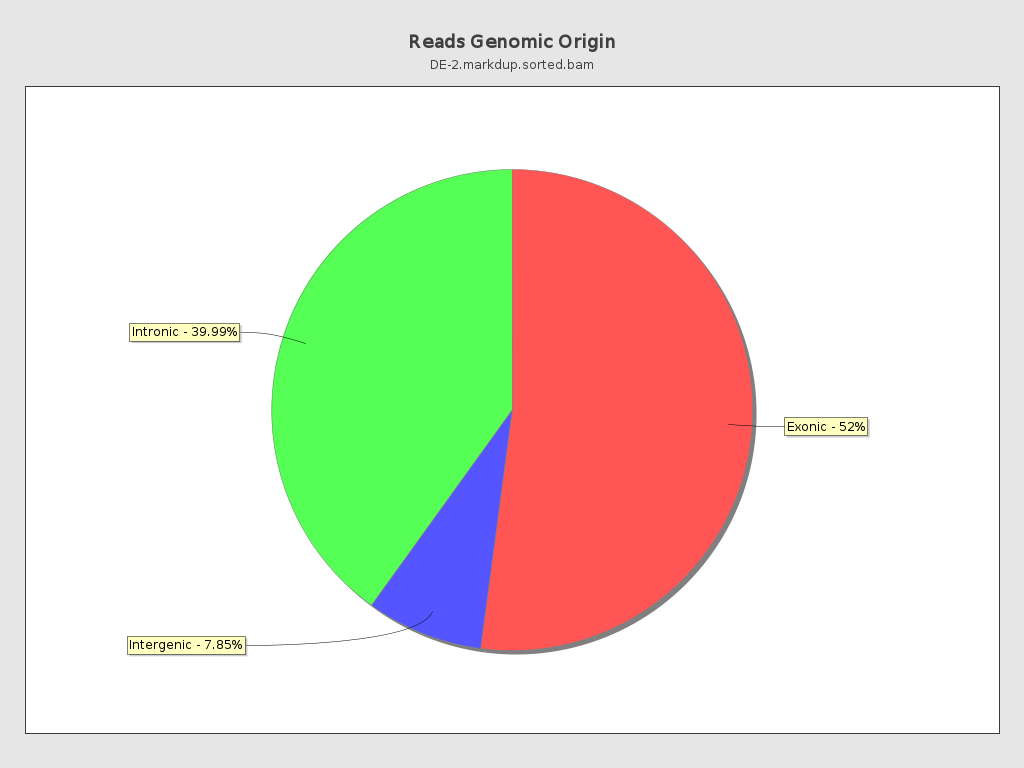
Reads genomic origin
| Exonic: | 82,091,802 / 52.16% |
| Intronic: | 62,939,845 / 39.99% |
| Intergenic: | 12,346,306 / 7.85% |
| Intronic/intergenic overlapping exon: | 4,385,610 / 2.79% |
Transcript coverage profile
| 5' bias: | 0.55 |
| 3' bias: | 0.34 |
| 5'-3' bias: | 1.3 |
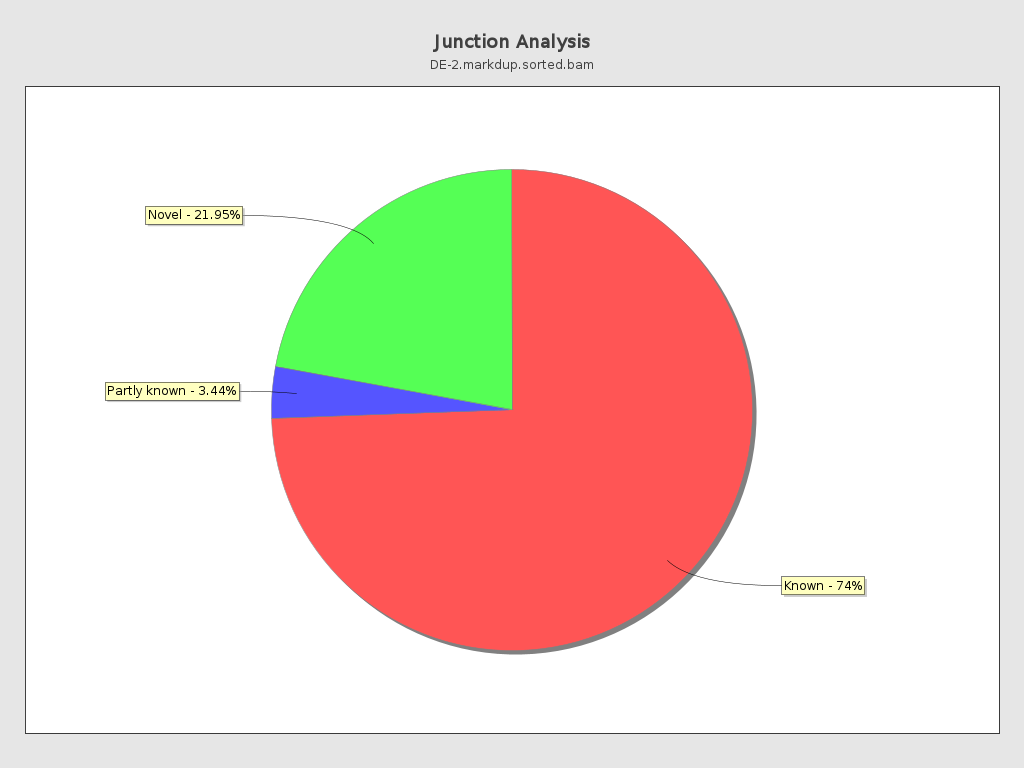
Junction analysis
| Reads at junctions: | 46,959,895 |
| ACCT | 5.61% |
| AGGT | 4.94% |
| TCCT | 3.53% |
| AGGA | 3.5% |
| ATCT | 3.09% |
| AGCT | 3.04% |
| GCCT | 2.7% |
| AGGC | 2.64% |
| CCCT | 2.48% |
| AGAT | 2.29% |
| AGGG | 2.26% |
.png)
.png)
.png)